
The high-stakes environment of modern drug discovery often feels like navigating a vast, multidimensional maze where a single structural misstep can derail years of intensive laboratory research. Pharmaceutical scientists frequently grapple with the immense difficulty of predicting how a prospective drug molecule will bind to its biological target, especially when the precise three-dimensional structure of that target remains an unsolved mystery. Optibrium has addressed this fundamental challenge by launching a sophisticated graphical user interface for its QuanSA technology, effectively bridging the divide between high-level computational modeling and the daily practicalities of medicinal chemistry. By integrating this powerful tool directly into the PyMOL molecular visualization system, the company provides researchers with a streamlined workflow that simplifies lead optimization and reduces the reliance on costly, iterative laboratory synthesis. This advancement transforms a once-cluttered landscape of data into a clear, actionable roadmap for identifying viable pre-clinical drug candidates with greater speed and precision.
Advancing Predictive Accuracy through Specialized Modeling
Technical Foundations: Quantitative Surface-Field Analysis
The core of this technological leap lies in Quantitative Surface-Field Analysis, or QuanSA, which serves as a specialized ligand-based binding affinity prediction tool within the BioPharmics 3D platform. Traditional modeling programs often hit a wall during the early stages of drug discovery because they require high-resolution imagery of the target protein, such as an X-ray crystal structure, to generate any useful data. In contrast, QuanSA is designed to thrive in the absence of such “lock and key” visual information by deriving its predictions from the known activities and structures of small molecules that have already demonstrated an ability to bind to the target. This capability is particularly critical in the current research climate where many therapeutic targets are considered “undruggable” or lack the structural clarity needed for structure-based design. By focusing on the ligands themselves, the system allows researchers to build highly accurate models long before the biological target has been fully crystallized or visualized.
Moving beyond the limitations of “black box” artificial intelligence, QuanSA utilizes a physically motivated machine learning approach that emphasizes the fundamental forces governing molecular recognition. While many modern AI tools provide a prediction without explaining the underlying logic, this technology explicitly models the physical factors, such as electrostatic fields and steric interactions, that dictate how a molecule seats itself within a binding pocket. This rigorous focus on physical reality allows the system to achieve a level of predictive accuracy that was previously reserved for far more computationally demanding methods like Free Energy Perturbation. By accounting for the nuances of molecular interaction in a mathematically transparent way, the platform provides scientists with high-quality binding data that is both reliable and explainable. Consequently, research teams can gain deep insights into molecular behavior without the need for the immense processing power typically associated with high-end quantum mechanics or exhaustive simulations.
The Role: Physical Principles in Machine Learning
The integration of physical principles into the machine learning framework marks a significant departure from purely statistical methods that often struggle with novel chemical space. By grounding the algorithm in the laws of thermodynamics and molecular mechanics, QuanSA ensures that its predictions remain relevant even when the molecules being studied differ significantly from the training set. This structural integrity is vital for lead optimization, where chemists are often exploring entirely new scaffolds to improve the potency or safety profile of a candidate. The method effectively simulates the environment of the binding site by analyzing the surface fields of active molecules, creating a virtual map of the requirements for high-affinity binding. This approach allows the software to identify subtle electronic or spatial features that a standard statistical model might overlook, providing a more robust foundation for making critical decisions about which chemical modifications will yield the best results in the lab.
Furthermore, the physical modeling approach used by QuanSA helps to eliminate much of the “noise” that can plague ligand-based drug design. By focusing on the actual forces of molecular interaction, the technology can distinguish between genuine affinity drivers and coincidental structural features. This clarity is essential for medicinal chemists who need to know exactly which atoms are contributing to binding and which are merely taking up space. The ability to calculate these interactions rapidly means that predictive modeling can be applied to thousands of potential compounds in a fraction of the time required by traditional simulation techniques. This speed does not come at the expense of accuracy, as the physically motivated nature of the tool ensures that the results are consistent with observed experimental data. Ultimately, this creates a more dependable bridge between the digital prediction and the physical reality of the laboratory bench, fostering greater confidence in the design process.
Enhancing Accessibility and Visual Communication
Democratizing Tools: Computational Chemistry for Bench Chemists
For several years, the QuanSA technology operated primarily as a command-line tool, a format that naturally restricted its use to a small circle of specialists with advanced expertise in Linux environments and computational chemistry. This technical barrier often meant that the bench chemists who were actually responsible for synthesizing new molecules were separated from the predictive power of the modeling tools they needed most. The introduction of the PyMOL plugin represents a major shift toward democratization, embedding these sophisticated capabilities within the most widely used molecular visualization software in the life sciences. By meeting scientists in a software environment they already use daily, Optibrium has removed the steep learning curve and technical friction that previously kept high-end modeling siloed in specialized departments. This accessibility ensures that predictive data is no longer a static report but a dynamic part of the creative design process for every chemist on a project team.
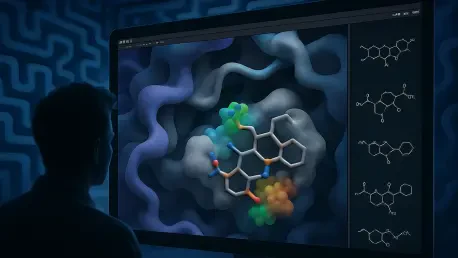
The graphical interface does more than just simplify navigation; it transforms abstract numerical data into detailed 3D representations that are far easier to interpret and act upon. Within the PyMOL environment, researchers can now see visual cues that represent complex molecular interactions in a highly intuitive manner. For example, the interface uses red and blue cones to indicate hydrogen bonding patterns and specific surface patches to highlight steric contributions, allowing scientists to see exactly where a molecule is clashing or fitting perfectly. This level of visual clarity enables a research team to understand why one compound might be ten times more potent than a structural twin that looks nearly identical on a 2D plane. By making these “invisible” forces visible, the plugin facilitates much more informed discussions between computational and medicinal chemists, ensuring that everyone is looking at the same structural challenges and opportunities through a clear, unified lens.
Bridging Interfaces: From Data Points to Design Decisions
The transition from raw data tables to an interactive visual interface represents a fundamental change in how drug discovery teams evaluate their chemical leads. In the past, a chemist might receive a list of predicted binding affinities and have to mentally reconstruct the 3D implications of those numbers, a process that is prone to human error and oversight. With the new PyMOL interface, the “why” behind every prediction is rendered in high definition, allowing for immediate feedback during the design phase. A researcher can virtually tweak a functional group and see how that change impacts the predicted surface interactions in real-time. This interactive loop accelerates the decision-making process, as it allows for the rapid rejection of ideas that are physically unfeasible and the prioritization of those that show the greatest promise. The interface effectively serves as a translator, turning the complex language of surface-field analysis into the visual language of structural chemistry.
Moreover, the democratized access provided by the plugin encourages a more collaborative and iterative approach to molecule design. When bench chemists can independently run QuanSA analyses within PyMOL, they are empowered to explore chemical variations that might not have been prioritized in a centralized modeling queue. This decentralization of expertise fosters innovation, as those closest to the synthetic challenges can immediately test the viability of their ideas against a physically accurate model. The result is a more agile research environment where the feedback loop between design and prediction is measured in minutes rather than days. By lowering the barrier to entry, Optibrium is not just providing a new tool; it is reshaping the culture of the research lab to be more data-driven and visually oriented. This integration ensures that sophisticated computational insights are a constant companion to chemical intuition, leading to more successful outcomes in the quest for new therapeutics.
Optimizing Resources and Strategic Integration
Economic Efficiency: Reducing the Synthesis and Testing Burden
In the current pharmaceutical industry, the gold standard for binding affinity prediction has long been Free Energy Perturbation, yet this method is notoriously resource-heavy, requiring massive computational clusters and significant amounts of time. QuanSA offers a validated alternative that delivers comparable results at a fraction of the hardware and time costs, which is a game-changer for budget-conscious research programs. By streamlining these calculations, the software allows research teams to significantly reduce the “synthesis and testing burden” that typically consumes the majority of a project’s budget. Instead of synthesizing hundreds of trial-and-error variations to find a single active lead, chemists can use the predictive insights of the plugin to focus their laboratory efforts only on the most promising candidates. This shift from physical iteration to digital refinement saves thousands of dollars in reagents and labor, while also shortening the overall timeline of the lead optimization phase.
The release of the QuanSA plugin is also a strategic move designed to consolidate Optibrium’s high-end modeling tools into a single, seamless workflow that aligns with broader industry trends. This launch follows the recent integration of the Surflex-Dock molecular docking tool into PyMOL, reflecting a clear commitment to creating a unified ecosystem for drug design. Modern biotech and pharmaceutical companies are increasingly moving away from fragmented software solutions that require constant data shuffling between incompatible platforms. By offering a suite of tools that function within a single, industry-standard interface, Optibrium ensures that its technology fits naturally into the existing routines of professional scientists. This strategic integration not only enhances user experience but also increases the likelihood that these advanced tools will be adopted early in the drug discovery lifecycle, where their impact on project direction and resource allocation is most profound.
Strategic Workflows: Integration for Maximum Impact
By offering this new plugin to current BioPharmics license holders at no additional cost, Optibrium is actively encouraging the widespread adoption of 3D modeling across the scientific community. This “low-barrier” strategy is intended to make sophisticated binding predictions a standard part of every project, regardless of the size of the company or the complexity of the biological target. When high-level modeling becomes ubiquitous, the entire industry benefits from more efficient resource usage and a higher success rate for pre-clinical candidates. This approach also allows researchers to tackle more challenging targets that were previously deemed too risky or difficult to model. As the cost of drug discovery continues to rise, tools that provide high-value insights with low overhead become indispensable. The integration of QuanSA into the PyMOL environment is a direct response to this need, providing a practical solution that balances scientific rigor with economic reality.
Furthermore, the focus on strategic integration helps to future-proof the research process against the increasing complexity of therapeutic modalities. Whether a team is working on small molecules, protein degraders, or other ligand-based therapies, having a flexible and accessible modeling platform is essential for staying competitive. The ability to run these analyses locally or on standard laboratory hardware without needing a dedicated supercomputer makes the technology highly scalable. This scalability ensures that as a project grows from initial screening to lead optimization, the same high-quality modeling tools can be used throughout the journey. By centralizing these capabilities within a familiar interface, the company is helping to build a more resilient and efficient research infrastructure. This strategic alignment of technology and workflow is key to accelerating the transition from laboratory discovery to clinical application, ensuring that promising new treatments reach patients more quickly.
Bridging the Gap Between Data and Intuition
Qualitative Insight: Beyond Numerical Affinity Predictions
The leadership at Optibrium has made it clear that the true value of the new PyMOL interface lies not just in the numbers it generates, but in the qualitative understanding it provides. While a numerical binding affinity prediction tells a scientist how strongly a molecule binds, the visual interface explains the “why” behind that strength, which is far more useful for making design interventions. VP Ann Cleves and CEO Matthew Segall emphasize that this qualitative insight allows for much more confident design decisions, as it enables researchers to visualize the specific molecular interactions that drive potency. This shift from raw data to visual insight represents a significant evolution in drug design, where the goal is to empower human scientists to apply their professional intuition alongside machine-generated predictions. By seeing the cones and patches of molecular force, a chemist can intuitively grasp the structural requirements of a binding site, leading to more creative and effective chemical solutions.
This unified approach to molecular design effectively bridges the gap between digital data and human expertise, creating a synergy that is more powerful than either component alone. When a scientist can see the physical reasons for a prediction, they are more likely to trust the results and act on them with confidence. This is particularly important when navigating the complex trade-offs inherent in drug discovery, such as balancing binding affinity with solubility or metabolic stability. The visual feedback provided by the QuanSA plugin allows for a more holistic view of the molecule, helping researchers to identify structural changes that improve one property without compromising another. By placing these capabilities into the hands of the wider scientific community, Optibrium is ensuring that the most advanced predictive modeling is accessible to those with the deepest understanding of the biological and chemical nuances of their specific projects.
Actionable Progress: The Path to Pre-Clinical Success
To capitalize on these advancements, research organizations should prioritize the integration of visual modeling tools into their early-stage lead optimization workflows. Rather than treating computational analysis as a separate, final step, teams should use the PyMOL interface as a primary design environment where chemical intuition and physical modeling interact in real-time. This proactive approach will allow for the immediate identification of structural liabilities and the rapid exploration of high-potency chemical space, ultimately streamlining the path to pre-clinical candidates. Scientists are encouraged to move beyond simple affinity scores and engage deeply with the visual surface-field data to gain a more nuanced understanding of their targets. By fostering this culture of visual and data-driven design, labs can maximize their synthetic resources and significantly improve the quality of the molecules they move forward into development.
Looking ahead, the success of drug discovery programs will increasingly depend on the ability to transform vast amounts of computational data into actionable medicinal progress. The democratization of expert-level tools like QuanSA through common interfaces represents a vital step in this direction. Research teams should look for opportunities to apply these ligand-based modeling techniques to targets where structural data is scarce, effectively expanding their reach into previously “undruggable” territory. As the industry continues to evolve, those who successfully merge the precision of physically motivated machine learning with the creative intuition of human chemists will be best positioned to deliver the next generation of therapeutic breakthroughs. The focus must remain on using these tools to drive better design decisions earlier in the process, ensuring that the transition from a digital model to a life-saving medicine is as efficient and predictable as possible.







