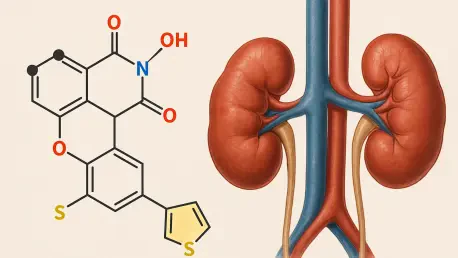

The biological machinery responsible for filtering waste sometimes malfunctions so catastrophically that routine metabolic processes turn into a source of systemic internal damage. Primary Hyperoxaluria represents a group of rare, inherited metabolic disorders that force the body into a state of chronic toxicity by overproducing oxalate, a compound that should ideally be excreted safely through the urinary tract. When this process fails, calcium oxalate crystals begin to precipitate within the delicate tissues of the kidneys, causing excruciating stones, persistent inflammation, and irreversible scarring. For those living with Primary Hyperoxaluria Type 2, or P##, the lack of targeted treatments has historically meant a rapid progression toward end-stage renal disease, often requiring invasive surgical interventions like dual liver and kidney transplants to preserve life in pediatric and young adult patients who are most frequently affected by this rare condition.
Targeted Inhibition: Blocking the Toxic Precursor Pathway
Recent breakthroughs from the Buck Institute for Research on Aging have introduced a small, orally administered molecule known as N-propargylglycine, which demonstrates a unique ability to halt this destructive process. The molecule functions by targeting a specific enzyme called hydroxyproline dehydrogenase, found within the mitochondria of the liver and kidneys. This enzyme typically facilitates the breakdown of hydroxyproline, an amino acid derived from collagen turnover, but in P## patients, this pathway becomes a primary source of excess oxalate production. By precisely inhibiting this enzyme, the new treatment effectively cuts off the chemical supply line before it can be converted into the toxic spillover that crystallizes in the renal system. This targeted approach prevents the initial formation of stones while simultaneously reducing the metabolic burden on the organ, offering a precision medicine solution that addresses the root cause rather than just managing symptoms.
The pharmacokinetics of this new molecule suggest that it is highly bioavailable, meaning it can be delivered effectively through a simple pill rather than requiring the frequent, painful injections or complex gene therapies often associated with rare metabolic disorders. Because the treatment focuses on a very specific metabolic route—the hydroxyproline-to-oxalate pathway—it minimizes the risk of off-target effects that often plague more broad-spectrum pharmaceutical interventions. Researchers have observed that the molecule easily penetrates the necessary tissues, ensuring that the therapeutic concentration reaches the liver and kidneys where the enzyme activity is most problematic. This ease of administration is a significant development for the roughly 1,700 patients currently diagnosed in the United States, as it provides a scalable and less invasive management strategy that could be integrated into daily life without the constant need for hospital-based care or extreme dietary restrictions.
Scientific Synergy: The Power of Cellular Resilience
The identification of N-propargylglycine as a renal therapy was not the result of a traditional nephrology project but emerged from an unexpected collaboration between experts in cancer and neurodegeneration. While investigating the molecule for its anti-tumor properties, scientists noticed that it significantly improved the health of cells by triggering a biological phenomenon known as mitohormesis. This process occurs when the body responds to a mild, controlled amount of mitochondrial stress by activating internal defense mechanisms that ultimately strengthen the cell’s resilience against future damage. The teams realized that the same pathway being studied to protect neurons in Huntington’s and Alzheimer’s diseases was directly linked to the metabolic defects seen in rare kidney disorders. This cross-disciplinary insight allowed the researchers to pivot their focus, applying their knowledge of mitochondrial health to solve a long-standing mystery in the field of renal medicine.
This dual-action mechanism distinguishes the new molecule from previous attempts at treating oxalate-related diseases, as it provides both offensive and defensive medical benefits simultaneously. On one hand, it stops the production of the harmful toxin, but on the other, it prepares the kidney’s internal structures to better survive any residual crystals that might still form. By “training” the mitochondria to be more robust, the therapy essentially creates a more durable organ that is less susceptible to the inflammation and cell death that typically follow crystal deposition. This emphasis on cellular resilience suggests that even in cases where complete elimination of oxalate is difficult, the kidney can maintain its functional integrity over long periods. This paradigm shift—from simply removing a toxin to actively fortifying the target organ—represents a sophisticated evolution in how researchers approach chronic and genetic diseases that involve localized tissue damage and systemic failure.
Empirical Results: Validation Through Rigorous Evidence
To confirm the efficacy of this discovery, the research team conducted extensive studies using mouse models that were genetically modified to simulate the lethal progression of human Primary Hyperoxaluria Type 2. In initial short-term trials lasting three weeks, the results were both immediate and profound, showing a drastic reduction in urinary oxalate levels and an almost total prevention of calcium oxalate stone formation. Pathological examinations of the kidney tissues revealed that the treated subjects maintained healthy renal architecture, with significantly less damage to the tubules compared to the control group. The untreated mice quickly developed severe injuries and crystal accumulations that mirrored the rapid decline seen in human patients. These findings provided the first clear evidence that the molecule could successfully interrupt the disease process in a living system, proving that the enzyme inhibition was effective enough to maintain normal physiological function despite the genetic defect.
Even more compelling evidence emerged from a long-term survival study where the subjects were placed on a diet specifically designed to accelerate the progression of the disease. While the untreated mice faced a median survival rate of only fifteen weeks due to catastrophic renal failure, the group receiving daily oral doses of the new molecule survived the entire twenty-four-week duration of the experiment. Remarkably, the physical health, body weight, and kidney function of the treated group remained indistinguishable from healthy wild-type subjects that did not have the metabolic disorder. This high level of protection suggests that the molecule is safe for long-term use and does not cause the adverse side effects often seen with potent enzyme inhibitors. The consistency of these results across different experimental phases solidified the molecule’s status as a top-tier candidate for clinical development, providing a clear proof-of-concept that could be translated into human trials.
Clinical Outlook: Future Perspectives on Renal Health
The broader implications of these findings suggest that the therapeutic reach of this molecule could extend far beyond the specific population of patients currently suffering from Primary Hyperoxaluria Type 2. Because Type 3 of the disease also relies on the same catabolic pathway in the liver to generate excess oxalate, scientists believe the treatment will be equally effective for those cases. This potential for a universal solution for multiple rare subtypes of the disorder could streamline the regulatory approval process and provide hope for thousands of families who have previously had no targeted options. Furthermore, the ability to strengthen mitochondrial health through mitohormesis could lead to new treatments for more common forms of kidney stones, which affect a significant portion of the global population. This approach provides a versatile platform for addressing various conditions where mitochondrial dysfunction and metabolic imbalances converge to cause progressive organ damage.
Moving forward, the scientific community focused on completing the necessary pharmacokinetic profiling and safety assessments to transition this research from the laboratory to human clinical settings. The discovery emphasized the critical importance of interdisciplinary research, demonstrating how insights from oncology and neuroscience could revolutionize the treatment of rare renal diseases. Medical experts recommended that future studies prioritize the development of standardized dosing protocols to maximize the mitohormetic benefits while ensuring consistent inhibition of the target enzyme. By establishing these clinical parameters, healthcare providers prepared to offer a transformative alternative to the invasive surgeries and transplants that once defined the standard of care. The successful validation of this molecule marked a definitive shift in the landscape of metabolic medicine, proving that targeted molecular therapies could restore function and provide a sustainable path toward long-term health for those previously facing a terminal diagnosis.