
What if the key to defeating some of humanity’s most devastating diseases lies within the tiniest boundaries of human cells, invisible to the naked eye, and only accessible through advanced technology? Membrane proteins, the unsung heroes of cellular function, act as gatekeepers, controlling the flow of nutrients and signals that keep life running. Yet, their elusive nature—tucked away in the greasy layers of cell membranes—has long baffled scientists, stalling progress in drug discovery for conditions like cancer. Now, a revolutionary computer-driven approach is tearing down these barriers, offering a glimpse into the atomic-level secrets of these vital structures. This breakthrough promises to reshape the landscape of medical research with unprecedented precision.
The Critical Role of Cellular Gatekeepers
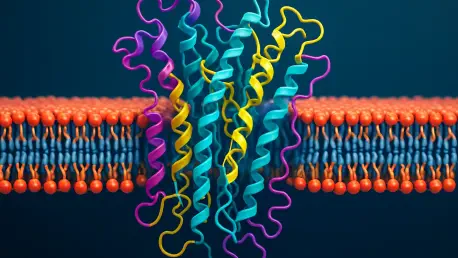
Membrane proteins are far more than mere components of a cell; they are the architects of life’s most fundamental processes. Embedded in the lipid bilayer, they manage everything from transporting essential molecules to enabling communication between cells. Their significance becomes starkly clear when they malfunction—such disruptions are linked to severe diseases, including cancer and neurodegenerative disorders, making them prime targets for therapeutic intervention. The pharmaceutical industry pours billions into developing drugs aimed at these proteins, recognizing their potential to unlock cures for millions.
However, the challenge of studying these proteins cannot be overstated. Their natural habitat within the lipid membrane renders them unstable when extracted, often collapsing traditional research methods. This obstacle has slowed advancements in understanding their structure and behavior, leaving critical gaps in knowledge. As the demand for precision medicine grows, finding innovative ways to probe these cellular gatekeepers has become an urgent priority for the scientific community.
A Computational Revolution in Protein Research
Enter a groundbreaking method pioneered by researchers at Scripps Research, where computer models are rewriting the rules of biological discovery. This approach sidesteps the instability of natural membrane proteins by designing synthetic versions from scratch, crafted to mimic their real-world counterparts. These digital creations, built using sophisticated software, provide a stable platform to explore intricate details that were once out of reach, offering a window into how these proteins fold and function within their native environment.
One striking focus of this research is a specific structural pattern known as the Gly-X6-Gly motif, a repeating sequence of glycine amino acids that appears every seven positions along a protein helix. This motif serves as a kind of molecular glue, stabilizing interactions between helical segments in the lipid membrane. Through computational simulations, scientists have confirmed that this pattern plays a pivotal role in maintaining the protein’s architecture, a finding that could redefine approaches to studying cellular mechanics.
Perhaps most surprising is the revelation of subtle forces at play within this motif. Weak hydrogen bonds, often dismissed as insignificant, were found to form a powerful network when combined, anchoring the protein’s structure with unexpected resilience. Lab tests further validated these models, showing synthetic proteins enduring extreme conditions like boiling—a testament to the accuracy and potential of this digital-first strategy.
Voices from the Cutting Edge
Behind this innovation stand dedicated minds like Marco Mravic, an assistant professor at Scripps Research, who sees this method as a game-changer. “This technology accelerates our ability to decode membrane proteins, closing the divide between theoretical insights and real-world treatments,” Mravic notes, highlighting the practical impact of their work. His enthusiasm reflects a broader shift in science toward embracing computational tools as essential allies in tackling biology’s toughest puzzles.
Equally compelling is the perspective of Kiana Golden, the first author of the study and the developer of the software driving these discoveries. “Watching our synthetic proteins fold exactly as the models predicted in the lab was a moment of pure validation,” Golden shares, underscoring the thrill of seeing digital predictions come to life. Their combined efforts, backed by rigorous experimentation, align with a growing consensus in the field that such approaches are not just viable but vital for future breakthroughs.
This synergy of computation and experimentation offers a model for collaboration, blending technical innovation with tangible results. As these tools become more refined, they promise to democratize access to complex research, enabling scientists worldwide to build on these findings and push the boundaries of what’s possible in understanding cellular function.
Harnessing Digital Tools for Drug Discovery
The implications of this computational leap extend far beyond academic curiosity, offering a clear roadmap for drug development. By creating synthetic membrane proteins, researchers can now test theories and observe interactions without the limitations posed by natural proteins’ fragility. This method allows for rapid prototyping of protein structures, providing a sandbox to explore potential therapeutic targets with unprecedented detail.
A strategic focus on structural motifs like Gly-X6-Gly equips scientists to pinpoint stabilizing mechanisms that could be exploited in drug design. Identifying these patterns helps reveal how mutations disrupt protein function, guiding the creation of molecules tailored to correct such flaws. This targeted approach could transform the way treatments are developed, moving away from broad-spectrum drugs toward personalized solutions rooted in molecular precision.
Integrating these insights into pharmaceutical pipelines holds immense promise. By mapping the atomic interactions of membrane proteins, researchers can design compounds that directly address disease-causing anomalies, potentially cutting years off drug development timelines. This framework not only enhances efficiency but also opens doors to therapies for conditions previously deemed untreatable, marking a new era in medical innovation.
Building on a Legacy of Discovery
Looking back, the journey to decode membrane proteins through computational models stands as a testament to human ingenuity overcoming nature’s toughest barriers. Scientists faced the daunting instability of these cellular gatekeepers head-on, crafting synthetic proxies that illuminated their hidden structures. Each discovery, from the stabilizing power of the Gly-X6-Gly motif to the unexpected strength of weak hydrogen bonds, added a vital piece to the puzzle of cellular function.
The path forward demands continued investment in computational tools, urging researchers to refine software and expand synthetic modeling capabilities. Collaboration across disciplines emerges as a cornerstone, with the potential to accelerate breakthroughs in drug design over the coming years, from 2025 to 2030. By scaling these efforts, the scientific community could target a wider array of diseases linked to membrane proteins, inching closer to cures.
Beyond immediate applications, this work inspires a broader reflection on how technology can unravel biology’s deepest mysteries. The challenge now rests in ensuring these innovations reach labs and clinics worldwide, translating digital insights into tangible health outcomes. With sustained effort, the legacy of this research promises to redefine medicine, one atomic interaction at a time.