
The pharmaceutical industry is currently witnessing a transformative shift in drug development as researchers move beyond the limitations of traditional small-molecule inhibitors toward the sophisticated field of Targeted Protein Degradation. While conventional therapies often focus on blocking the active sites of problematic proteins, this approach frequently encounters obstacles such as competitive inhibition and the requirement for high drug concentrations that can lead to systemic toxicity. Targeted Protein Degradation offers a more comprehensive solution by utilizing the cell’s internal machinery to physically eliminate disease-causing proteins rather than simply trying to suppress their function. This methodology was a primary focus at the ELRIG 2025 meeting, where scientific leaders discussed how chemical rewiring could expand the reach of modern medicine to include previously undruggable targets. By transitioning from protein blocking to protein destruction, the medical community is opening new doors for treating complex conditions that have long resisted standard pharmacological interventions.
Understanding the Cellular Disposal Machinery
The Mechanics: Protein Recycling
The internal health of a cell is maintained by the Ubiquitin-Proteasome System, which functions as a sophisticated waste management network responsible for degrading damaged or unnecessary proteins. At the center of this biological process are E3 ligases, a specialized class of enzymes that act as molecular quality control officers by identifying specific substrates destined for elimination. Once a target protein is recognized, the E3 ligase facilitates the attachment of a small regulatory protein called ubiquitin, which serves as a biochemical tag. This tagging process essentially marks the protein for delivery to the proteasome, a massive protein complex that acts as an industrial-scale shredder. Inside the proteasome, the tagged protein is broken down into its constituent amino acids, which the cell can then recycle to build new structures. This elegant recycling system ensures that the cellular environment remains free of toxic accumulations and functional aberrations.
The Challenge: Overcoming Technological Bottlenecks
Despite the human genome containing more than 600 distinct E3 ligases, the current generation of protein-degrading drugs relies almost exclusively on a tiny fraction of this diversity, specifically Cereblon and Von Hippel–Lindau. This heavy reliance on just two ligases presents a significant bottleneck for drug discovery, as it limits the types of tissues that can be effectively treated and increases the risk of therapeutic resistance. If a cancer cell loses the ability to express these specific ligases or develops mutations that prevent drug binding, the entire treatment strategy becomes ineffective. Expanding the toolkit to include a wider variety of the 600 available E3 ligases is essential for creating the next generation of precision therapies. By unlocking new ligases, researchers can design drugs that are highly tissue-specific, ensuring that protein degradation only occurs where it is medically necessary, thereby reducing the likelihood of off-target effects and improving overall patient outcomes in complex clinical cases.
Learning from Viral Strategies
The Strategy: Viruses as Blueprints
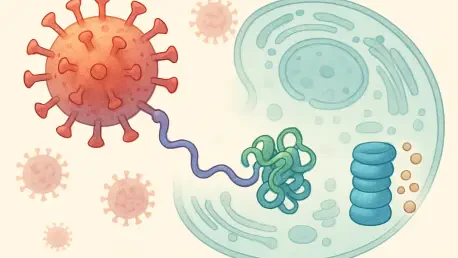
Nature has already provided a remarkably efficient blueprint for expanding the reach of protein degradation through the evolutionary tactics of various viral pathogens. Over millions of years, viruses have developed the ability to survive within host organisms by hijacking human E3 ligases to destroy antiviral proteins that would otherwise block viral replication. This natural form of “chemical rewiring” allows a virus to redirect a host enzyme toward a target it was never originally intended to encounter. By studying these viral-host interactions, scientists are gaining critical insights into which E3 ligases are naturally susceptible to being repurposed for therapeutic goals. This approach suggests that if a virus can successfully manipulate a human enzyme to eliminate a defense protein, medical researchers can employ similar logic to design synthetic molecules that direct the same enzymes toward proteins responsible for human diseases. This bio-inspired strategy represents a significant departure from traditional drug design.
The Methodology: The Search for Candidates
To identify new candidates for protein degradation, researchers at IRB Barcelona implemented a rigorous computational pipeline designed to sift through the human ligasome for high-priority targets. The team specifically looked for E3 ligases that possessed high-resolution structural data and a documented history of being manipulated by viral proteins, which indicated their potential for therapeutic hijacking. From an initial pool of over 600 candidates, the search was eventually narrowed down to five high-potential ligases, with a substrate receptor known as FEM1C emerging as a primary lead for further development. FEM1C is particularly attractive because it features a distinct surface pocket that is structurally accessible to small-molecule drugs. While viruses typically use large peptides to engage this pocket, the modern challenge involves engineering much smaller, drug-like molecules that can achieve the same result. The identification of FEM1C proves that systematic screening can uncover new molecular handles for the disposal of disease-linked proteins.
The Path to Precision Medicine
The Discovery: Developing Potent Molecular Binders
The transition from identifying a target to developing a viable drug required extensive experimental validation and the screening of massive chemical libraries to find effective molecular binders. Researchers sought molecules that could bind to the FEM1C pocket with high affinity, eventually identifying a series of potential ligands that demonstrated the necessary chemical properties. Through structure-driven optimization, the team developed a lead compound designated as C41, which exhibits nanomolar affinity for its target. This high level of potency means that C41 can effectively engage the FEM1C ligase even at extremely low concentrations, making it an ideal component for the construction of Proteolysis-Targeting Chimeras. These bifunctional molecules act as a bridge, bringing the E3 ligase into close proximity with a disease-causing protein to initiate the degradation process. The creation of such potent binders is a critical milestone, as it provides the modular components needed to target a diverse array of previously inaccessible pathological proteins.
The Implementation: A New Era of Design
The successful targeting of FEM1C established a proof-of-concept for a more rational and scalable approach to drug discovery that moved away from the accidental findings of the past. By integrating computational biology, structural analysis, and the evolutionary lessons learned from viral hijacking, the scientific community began building a robust platform for the future of precision medicine. This systematic expansion of the degradable proteome ensured that the cell’s own recycling machinery could be directed with surgical accuracy toward virtually any protein in the human body. As more E3 ligases were unlocked and characterized, the ability to treat chronic and complex diseases with high specificity became a practical reality rather than a theoretical goal. The medical community looked toward establishing standardized protocols for ligase recruitment, which allowed for the rapid development of therapies tailored to the unique genetic and cellular profiles of individual patients. This evolution in biotechnology transformed the proteasome into a versatile tool for modern pharmacology.