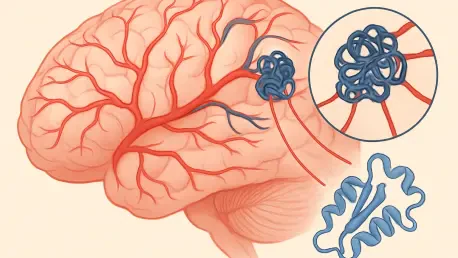

Deep within the intricate labyrinth of the human brain, a silent and often invisible architect may be weaving a cluster of fragile, mulberry-shaped vessels that threaten to rupture at any given moment. These structural anomalies, known as Cerebral Cavernous Malformations (CCMs), are far more common than many realize, potentially affecting one out of every two hundred individuals. Unlike healthy blood vessels that facilitate a steady and secure flow of oxygenated blood, these malformations are composed of thin-walled, dilated channels that lack the necessary structural integrity to withstand normal physiological pressures. This inherent fragility makes them prone to slow leakage or sudden, catastrophic rupture, which can transform a routine day into a neurological emergency within seconds.
The physical presence of these “mulberries” within the central nervous system creates a precarious environment for the surrounding brain tissue. When these vessels fail, the resulting intracranial hemorrhage can manifest as a debilitating stroke, a sudden onset of focal neurological deficits, or chronic, treatment-resistant seizures. For those diagnosed with the condition, the psychological burden is often as heavy as the physical risk. Living with an abnormality that can bleed at any time creates a state of perpetual anxiety, especially since many of these lesions are discovered incidentally during unrelated imaging. The medical community has long recognized the danger these structures pose, yet the biological mechanisms that drive their growth remained partially obscured, leaving clinicians with a narrow set of tools to combat the progression of the disease.
The High Stakes of Vascular Fragility and Limited Intervention
The true severity of Cerebral Cavernous Malformations is found in their clinical unpredictability and the profound lack of non-invasive therapeutic options. While the primary cause of these lesions is rooted in specific genetic mutations, the actual physical manifestation is characterized by a catastrophic breakdown of the endothelial lining that keeps blood contained within the vascular system. For decades, the standard of care for symptomatic patients has been remarkably limited, revolving almost entirely around the invasive removal of the affected tissue. This creates a difficult choice for patients and surgeons alike, as many lesions are situated deep within the brainstem or in eloquent areas responsible for speech and movement, making surgery a high-stakes gamble with the potential for permanent disability.
This clinical gap has left a significant portion of the patient population in a state of “watchful waiting,” a period of time where the only action taken is periodic imaging to check for lesion growth or recent hemorrhage. This passive approach highlights an urgent need for a therapeutic breakthrough that moves the focus from the scalpel to the pharmacy. If researchers could identify the specific molecular triggers that cause these vessels to malform and leak, it would be possible to develop a pharmacological intervention that stabilizes the brain’s vasculature before a crisis occurs. Transitioning away from reactive surgery toward proactive prevention is the primary frontline in the battle against neurological catastrophe for millions of people worldwide who carry these genetic markers.
Decoding the Molecular Chain Reaction: The Path to Dysfunction
The development of a cavernous malformation is not a random event but rather the end result of a complex signaling “domino effect” occurring within the endothelial cells. Modern molecular biology has identified the MEKK3-KLF2/4 signaling pathway as the primary engine for this vascular dysfunction. In patients with the necessary genetic mutations, this specific cascade becomes hyperactive, sending a constant stream of signals that tell the blood vessel cells to grow and reorganize in a chaotic, dysfunctional manner. This pathway acts as the foundational trigger, setting the stage for the physical breakdown of the vessel walls and the eventual formation of the characteristic mulberry-shaped clusters.
A critical component of this growth process is the phosphoinositide 3-kinase (PI3K) enzyme, which is stimulated by the initial hyperactivation of the MEKK3-KLF2/4 pathway. While PI3K is the actual driver of the lesion growth, it presents a significant therapeutic dilemma. Because this enzyme is vital to a wide range of general health functions, including the regulation of metabolism, cell growth, and the immune system, blocking it systemically causes severe and often intolerable side effects. In previous experimental trials, while inhibiting PI3K successfully stopped the growth of CCMs, the resulting toxicity made the treatment unfeasible for long-term human use. This led researchers to look for a more precise point of intervention that could provide the same benefits without the systemic fallout.
The breakthrough came with the identification of the TIE2 protein as the “missing link” that connects the upstream genetic mutation to the downstream PI3K growth signal. TIE2 is a receptor protein located on the surface of endothelial cells, and it appears to act as a localized bottleneck for the entire signaling process. By focusing on TIE2, scientists discovered that they could disrupt the signal specifically within the vascular system without interfering with the broad metabolic roles played by PI3K in other parts of the body. This discovery transformed TIE2 into a highly attractive target for precision medicine, offering a way to stop the disease at its molecular source.
Expert Insights and the Rebastinib Discovery: A New Paradigm
Groundbreaking research led by the Perelman School of Medicine at the University of Pennsylvania has recently shifted the entire paradigm of how these malformations are understood and treated. Dr. Mark L. Kahn and his team conducted a series of sophisticated experiments which revealed that TIE2 levels are significantly elevated in the specific endothelial cells surrounding CCM lesions. This elevation is not merely a side effect but a critical driver of the disease’s progression. By utilizing advanced imaging and molecular analysis, the team was able to prove that the genetic mutations responsible for CCM directly lead to an overabundance of TIE2, which then triggers the harmful PI3K signaling cascade.
To test the viability of TIE2 as a therapeutic target, the researchers utilized rebastinib, an oral small-molecule drug that was originally designed as a TIE2 inhibitor for other applications. In experimental models designed to replicate the human version of the disease, the administration of rebastinib demonstrated remarkable preventative success. The study showed that by blocking the activity of the TIE2 receptor, the team could successfully prevent the development of new vascular malformations and stop the expansion of existing ones. This success suggested that the disease could be managed through a targeted pharmacological approach, rather than relying solely on the hope that a lesion does not rupture before it can be surgically addressed.
This discovery represents a significant shift in focus toward the receptor proteins on the cell surface. By targeting TIE2, the research suggests a method for neutralizing the disease’s growth signals before they can activate the more problematic and generalized enzymes within the cell. This “endothelial-centered” strategy avoids the broad-spectrum toxicity that doomed previous drug candidates, offering a more refined and tolerable path forward for patients who may require lifelong therapy to maintain the stability of their brain’s blood vessels.
A New Framework for Chronic CCM Management: Moving Toward the Clinic
The identification of TIE2 as a viable target provides a clear and actionable roadmap for moving discovery from the laboratory into clinical practice. By focusing on this specific gatekeeper, physicians can look toward a future where “vascular stabilization” is a standard part of neurological care. This strategy allows for a high degree of precision, as the therapy targets the specific receptors that are overactive in the disease state while leaving the rest of the body’s systems largely unaffected. This transition from invasive surgery to chronic, manageable pharmacotherapy could redefine the prognosis for patients who currently face a lifetime of uncertainty.
The availability of small-molecule inhibitors like rebastinib is particularly promising because it opens the door for a pill-based regimen. For patients with the familial form of the disease, who are genetically predisposed to developing multiple new lesions throughout their lives, a proactive, oral treatment could offer a way to stabilize the brain’s vasculature permanently. Instead of waiting for a lesion to reach a critical size or cause a hemorrhage, these individuals could potentially take a daily medication that prevents the malformations from ever forming. This proactive stance would drastically reduce the cumulative risk of strokes and seizures, fundamentally changing the life trajectory for entire families carrying these genetic mutations.
Ultimately, the goal of this pharmacological approach is to eliminate the primary cause of CCM-related catastrophes before they manifest as clinical symptoms. By preventing the formation of fragile, leaky clusters, the medical community can move toward a model of care that prioritizes prevention and long-term management. This shift not only promises to save lives but also to preserve the quality of life for those who would otherwise be subjected to the risks of neurosurgery or the constant threat of intracranial bleeding. As this research progresses into human clinical trials, the prospect of a world where brain vascular malformations are a preventable condition is closer than ever before.
The research team at the University of Pennsylvania successfully identified the molecular bridge that allowed genetic mutations to trigger the growth of dangerous brain lesions. This work established TIE2 as a critical bottleneck in the signaling pathway, providing a localized target that avoided the systemic toxicity of earlier drug candidates. By utilizing rebastinib in experimental models, the scientists demonstrated that pharmacological intervention could effectively halt the progression of the disease. These findings provided a foundation for transitioning away from high-risk surgical procedures toward a preventive, oral treatment strategy. This milestone shifted the clinical focus toward long-term vascular stabilization and offered a potential solution for patients living with the constant risk of intracranial hemorrhage.